Environment & Energy
Related: About this forumKinetics Over Extended Periods of Solvated Electrons in the Remediation of PFAS and Halocarbons.
I envision a system in which our energy production cleans up, via continuous flows of the contaminated media - air and water, and by extraction, indirectly, land - the pollution left by our generation. I'm not trying to be glib, there is a long and extremely difficult - and possibly expensive path to such a world - but it strikes me as just on the edge of feasibility.
Among the most serious pollutants are the halocarbons, with the most actively discussed set of compounds being the PFAS, per(poly)fluorinated alkyl substances.
I discussed the radiolytic degradation of these a little over two years ago in this space: A Nice Scientific Review Article on the Destruction of Persistant Perfluoroorganic Pollutants.
The system I discussed then was really relevant to destruction in processed water, which is not entirely unrealistic in a scenario where solid phase extraction/elution is utilized to capture the PFAS from a natural matrix such as river water, groundwater or seawater. My vision for this kind of system would be to elute the contaminants into highly radioactive water whereupon they would be mineralized into a fluorite analog - fluorite is the mineral CaF2 - BaF2 is similar.
I came across a paper tonight that discusses the properties of electrons in natural (unprocessed) water, this one: Quantifying Hydrated Electron Transformation Kinetics in UV-Advanced Reduction Processes Using the Re–,UV Method Benjamin D. Fennell, Adam Odorisio, and Garrett McKay Environmental Science & Technology 2022 56 (14), 10329-10338.
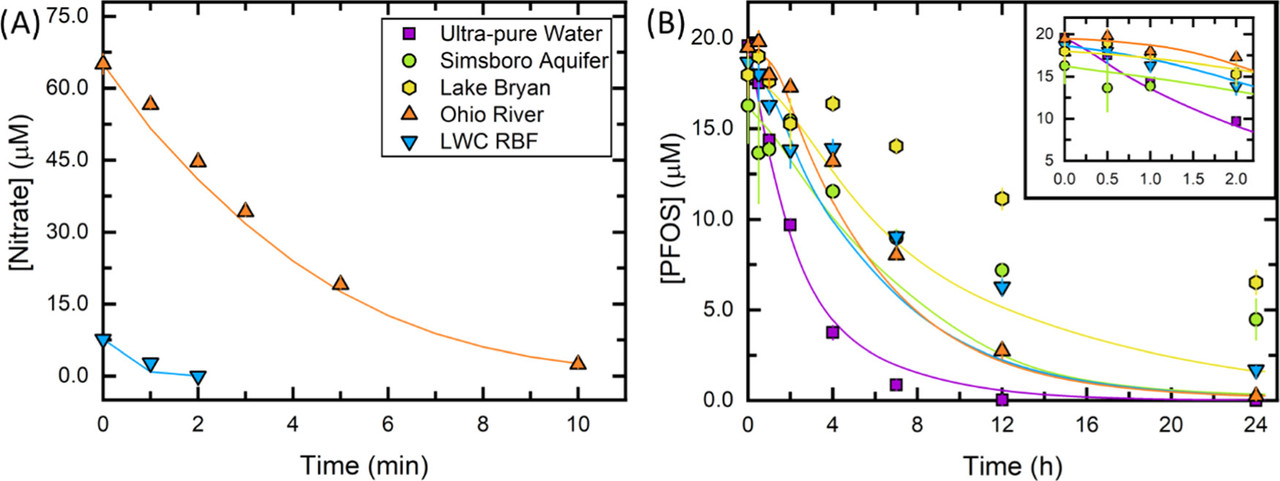
Regrettably I don't have a lot of time to discuss this paper in a lot of detail, but perhaps a brief excerpt and some pictures will give the flavor. MCAA here is a model compound, monochloroacetic acid. Chlorinated carbons are also a serious matter; although they've been banned largely, we still have huge quantities of many of these remaining, DDT and PCBS for just two examples.
The introduction to the paper:
Quantifying the time-based hydrated electron concentration ([eaq–]t) is important in UV-ARP treatment due to the varied treatment times required for contaminant degradation and the potential for eaq– formation and scavenging conditions to change over these time scales. For instance, nitrate, which has high reactivity with eaq–, completely degrades on the time scale of minutes in UV-ARP systems, (2,28,29) whereas degradation half-lives of hours are typical for more recalcitrant contaminants like PFAS. (15,22) It is likely that the [eaq–]t experienced by nitrate and PFAS at early treatment times is not the same as at later treatment times. This contrasts with UV-AOP in which hydroxyl radical (•OH)-mediated contaminant transformation kinetics occur on much shorter time scales, (30) such that the [•OH] in these systems is assumed to be at a steady state. The [eaq–]t available for contaminant degradation is impacted by eaq– scavengers present in water. Past studies have demonstrated the inhibitory impacts eaq– scavengers such as dissolved oxygen, (6,31) nitrate, (10) dissolved organic matter (DOM), (8,11) and bicarbonate (11) have on contaminant degradation. While these studies highlight the importance of individual eaq– scavengers, the combined, long-term impact of eaq– scavengers most often present in natural waters has not been considered.
The purpose of this paper is to develop a method for characterizing the key properties of eaq–-mediated transformation kinetics in UV-ARP, namely, [eaq–]t and kS,t′ (eaq– scavenging capacity). To this end, we deployed a probe compound selective for eaq– in the UV/sulfite system, monochloroacetate (MCAA), and developed the Re–,UV method (defined as the eaq– exposure per UV fluence) in analogy to ROH,UV previously used for the UV/H2O2 system. (32) We then use the Re–,UV method to model the degradation rates of two contaminants─nitrate (NO3–) and perfluorooctane sulfonate (PFOS)─in four source waters with varying water quality. The results demonstrate that a water’s initial eaq– scavenging capacity does not necessarily determine the ultimate treatability of PFOS. Rather, the extent of PFOS degradation is determined by the total eaq– exposure as measured by the Re–,UV method...
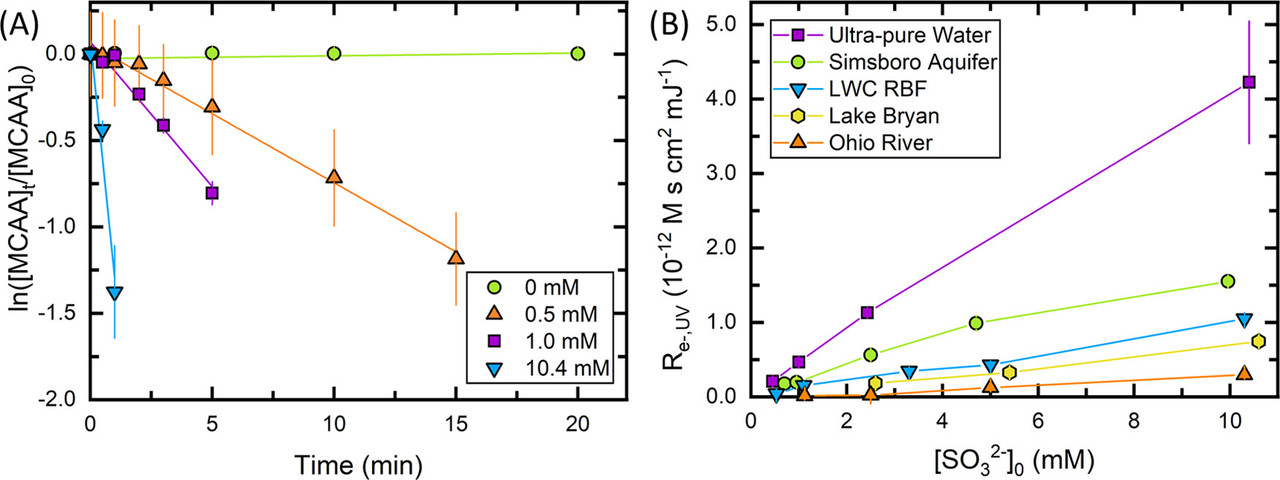
The caption:
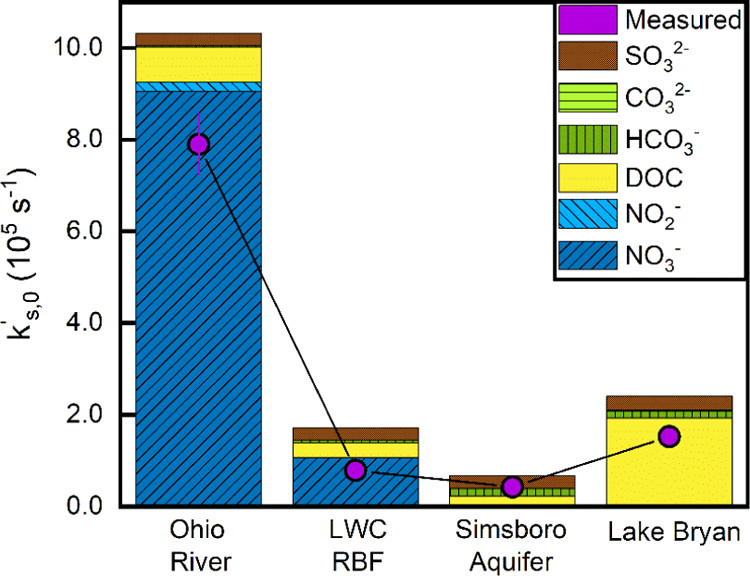
The caption:
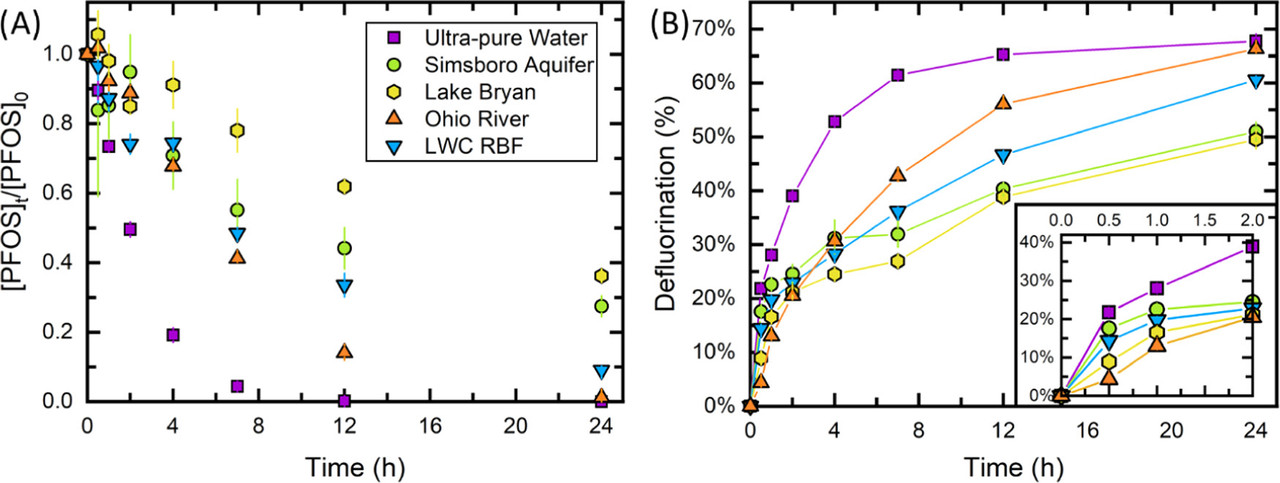
The caption:
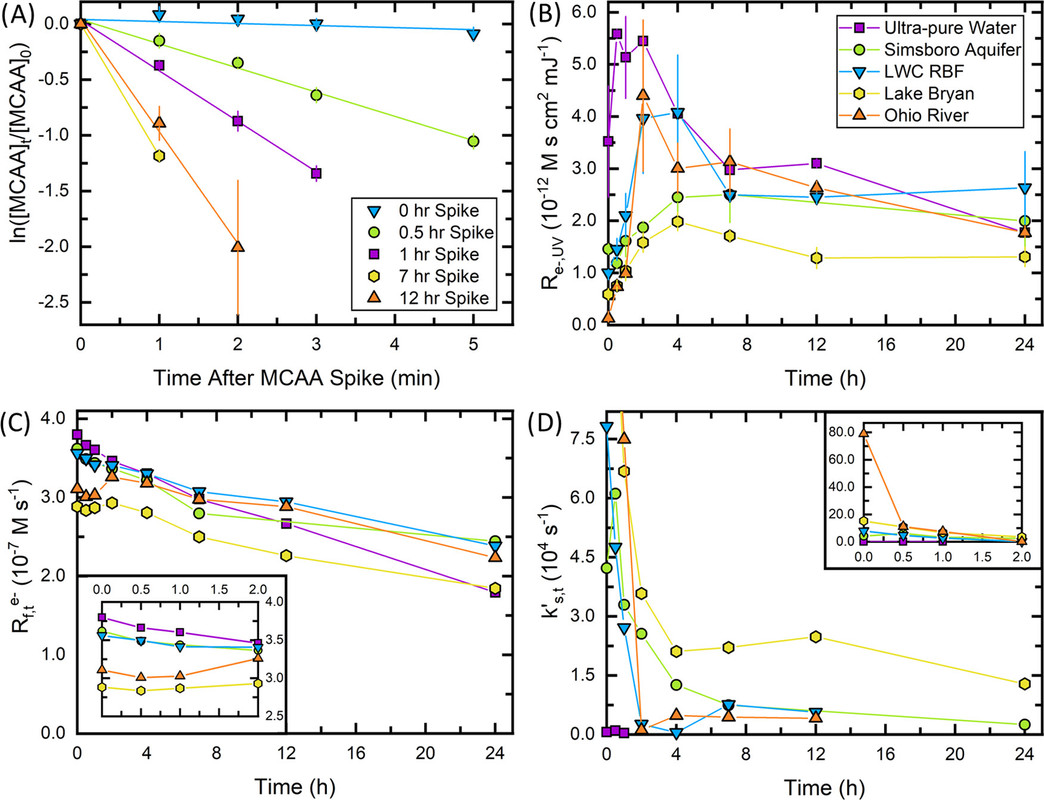
The caption:
Figure 4. Temporal variation in eaq– availability during a 24 h UV/sulfite experiment. (A) Measured MCAA degradation (symbols) as a function of time in Ohio River (20 μM [MCAA]0 spiked into reactor prior to each time point). The best linear fit (line) is also shown. (B) Re–,UV, (C) Rf,teaq–, and (D) kS,t′ (eq 3.6) as function of time for all waters (20 μM [MCAA]0 spiked into reactor prior to each time point). Legend in panel (B) applies to panels (C) and (D). Reported kS,t′ values in panel (D) are within the eaq– scavenging capacity measurement limit established by the probe compound. All experiments include the following conditions: 10 W low-pressure Hg lamp, pH0 = 9.4–9.7, 20 °C, [MCAA]0 spikes = 20 μM, [SO32–]0 = 10.0 mM, [PFOS]0 = 20.0 μM, and [borate] = 1.0 mM (ultrapure water only). All chemicals were spiked into the waters at time 0. Markers represent the mean of duplicate measurements and error bars represent the range between the duplicates.
The conclusion:
Our results suggest that high alkalinity may inadvertently increase the treatment time or amount of sensitizer chemical required for complete PFOS degradation when applying UV-ARP treatment to groundwaters. Furthermore, DOC was shown to both increase the eaq– scavenging capacity by having a moderately fast eaq– bimolecular rate constant (∼108 MC–1s–1) and decrease Rf,teaq– by shielding sulfite from incoming photons. The role of DOC in eaq– scavenging deserves further attention. For example, we observed that Lake Bryan (with an initial [DOC] of almost 10 mgC L–1) exhibited a higher eaq– scavenging capacity than other source waters after ∼12 h of treatment. This is particularly relevant for UV-ARP treatment of concentrated PFAS sources such as ion exchange regenerate and reverse osmosis concentrate. Effective UV-ARP treatment in waters with high DOC may require pretreatment to lessen the impacts of this eaq– scavenger.
These kinetics suggest that these systems will not work well, except through multiple passes in continuous flow systems, and that the solid phase extraction/elution system might be better, except in situations where water is being conditioned for use, such as in the case of drinking water restored from contaminated bodies of water.