Electrochemical Molecular Switches for the Capture and Release of Uranium. [View all]
The paper I'll discuss in this post is this one: Redox-switchable carboranes for uranium capture and release (Gabriel Ménard et al, Nature volume 577, pages 652–655(2020))
According to a DOE report, DOE/EM-0275 as of 1996, the US government had in stock about 585 million kg of depleted uranium, beyond the 25 million kg enriched to about 3% in U-235, about 610 million kg.
A kg of plutonium, the starting material for which is depleted uranium, contains about 80.3 trillion joules of energy, completely fissioned.
The world was, as of 2018, consuming about 600 exajoules (600 million trillion joules) of energy each year.
Recently there have been efforts by a number of companies, one of the most well known being Bill Gates' Terrapower, to commercialize "breed and burn" type reactors that transmute depleted uranium into plutonium in situ.
It follows that this US inventory is sufficient, at current levels of energy demand to power all the world's energy for about 80 years, no dangerous natural gas, no dangerous petroleum, no coal mining for energy purposes. Of course, there are other uranium inventories elsewhere in the world. In addition, a side product of the useless wind industry and the electric car industry, both of which depend on access to iron neodymium boride magnets, often doped with dysprosium - lanthanides - is the radioactive element thorium. Collected from lanthanide mile tailings, dumps, in which the thorium has been partially refined, this thorium is also a valuable nuclear fuel. It is reasonable to say that in a "breed and burn" powered world it would be unnecessary to mine any fuels for several centuries.
Fracked rock, which has been eternally pulverized for a few decades of "good times" by all of us self declared "green" people also represents a potential source of uranium: The radon dumped by the gas industry in Pennsylvania's Reading Pronge gas fields indicates that this pulverized rock, over which water may flow for millenia upon millenia, is also a potential uranium source.
Finally, since that establishment of oxygen in the planetary atmosphere, a continuous uranium cycle has been established in the planetary oceans; they contain about 4.5 - 5 billion tons of uranium.
There has much discussion of refining uranium from dilute sources, seawater, run-off from uranium mine tailings, and natural uranium formations both for the purposes of obtaining fuel as well as to remediate areas of anthropomorphic contamination or natural uranium flows. Uranium is a chemotoxic element, notably having effects on renal and other tissues. Many thousands of papers on this subject have been published in the scientific literature; I almost certainly have hundreds in my personal electronic files. Many of these papers concern organic resins, notably amidoxime functionalized resins. There are also inorganic species that have been advanced for this purpose. What is of interest about this laboratory scale material is that it can more or less breathe uranium, in effect "inhale" and "exhale" it by the application of electrical currents.
From the introduction:
Closo and nido refer to something known as the "Wade-Mingo" rules, and refer to the presence of a complete platonic solid structure, in this case icosahedral symmetry, having all vertices represented, closo or one vertex missing, nido. (The symmetry of in these cases is not truly icosahedral, since the symmetry is "disturbed" or "degraded" by the presence of the functionalized carbon. The carbon in this boron hydride structure is functionalized with diphenylphosphine oxide.
Figure 1:
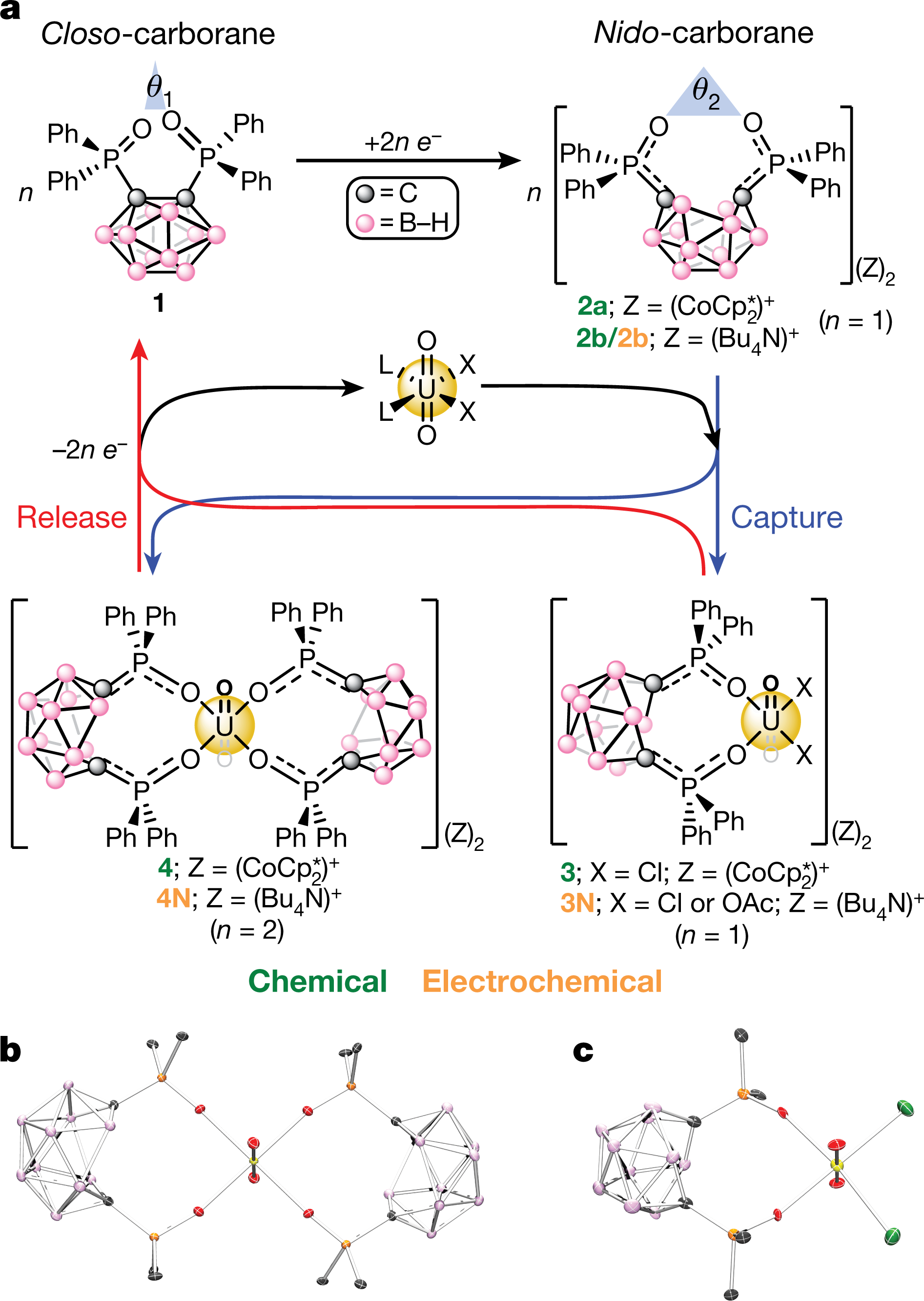
The caption:
a, General chemical or electrochemical mono- or bi-phasic capture of uranyl from UO2X2L2 (X = Cl−, OAc−; L = THF, Ph3PO) using the reduced ‘open’-cage nido-carboranes (2a/2b) generated by reduction (for example, CoCp∗2CoCp2∗ or negative bias) of the ‘closed’-cage closo-carborane (1). The corresponding relative bite angles (θ
 are also shown. Oxidation (for example, [FeCp2][PF6] or positive bias) of the captured products 3/4 or 3N/4N leads to UO22+ release. Compounds labelled in green have been chemically isolated, whereas compounds in orange are proposed electrochemical products (see Methods). Blue and red pathways represent UO22+ capture and release, respectively. b, c, Solid-state molecular structures of 4 (b) and 3 (c) obtained from XRD studies. H atoms, [CoCp∗2]+[CoCp2∗]+ counter cations, phenyl C–H linkages and all co-crystallized solvent molecules are omitted for clarity. See Extended Data Fig. 1for the structures of 1 and 2a.
are also shown. Oxidation (for example, [FeCp2][PF6] or positive bias) of the captured products 3/4 or 3N/4N leads to UO22+ release. Compounds labelled in green have been chemically isolated, whereas compounds in orange are proposed electrochemical products (see Methods). Blue and red pathways represent UO22+ capture and release, respectively. b, c, Solid-state molecular structures of 4 (b) and 3 (c) obtained from XRD studies. H atoms, [CoCp∗2]+[CoCp2∗]+ counter cations, phenyl C–H linkages and all co-crystallized solvent molecules are omitted for clarity. See Extended Data Fig. 1for the structures of 1 and 2a.Many of the experiments described in the full paper take place in organic solvents, which of course, is not seawater, but nevertheless the system is definitely quite interesting, and one can imagine modifications.
Anyway, the system operates electrochemically.
Figure 2:
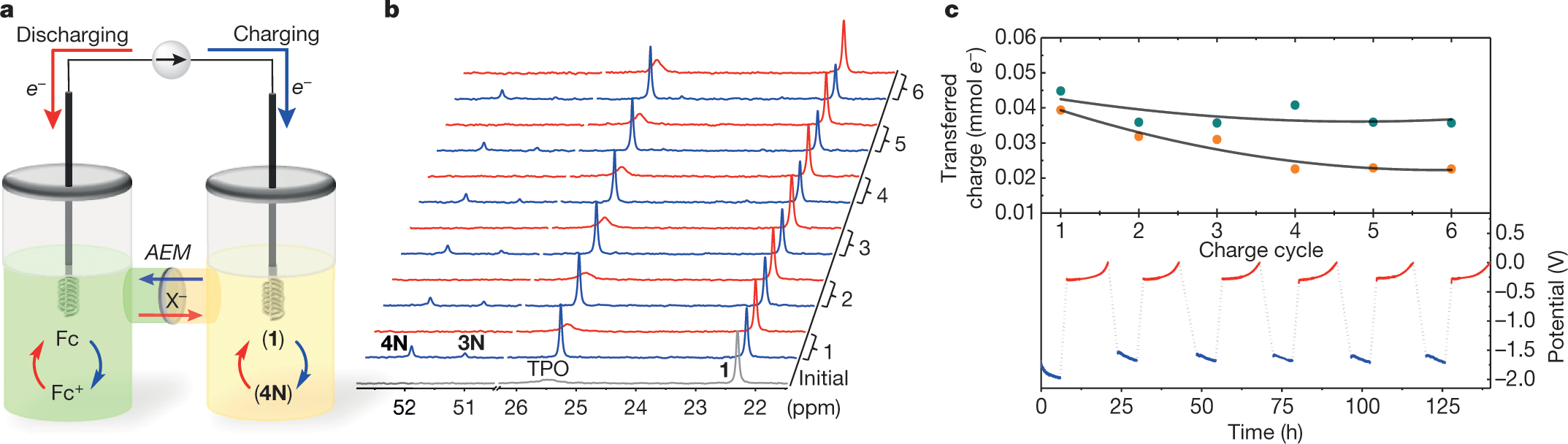
The caption:
The issue of organic solvents is addressed as shown in figure 3, which essentially is an extraction procedure.
Figure 3:
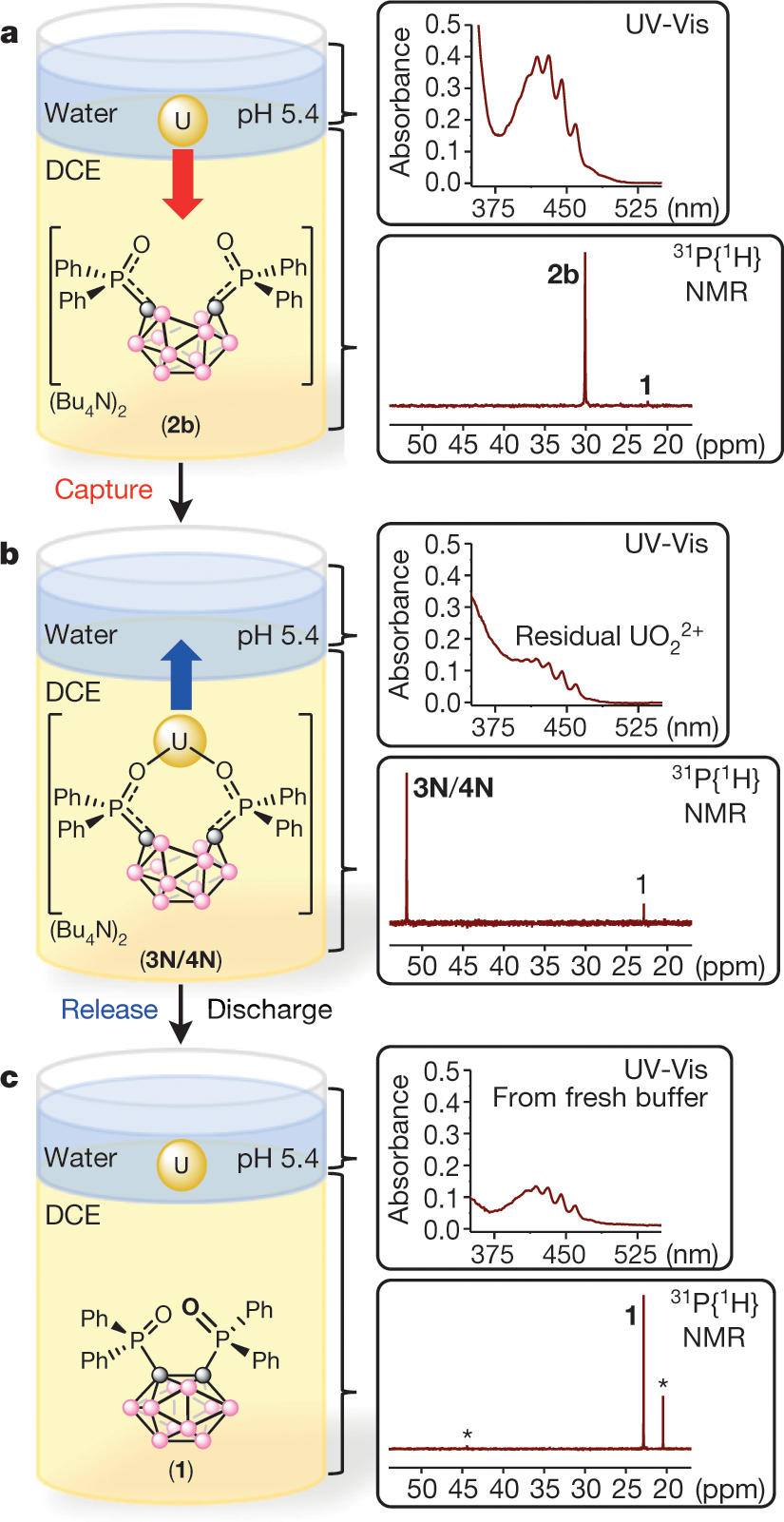
The caption:
. Inset, aqueous UV-Vis and organic 31P{1H} NMR spectra after reduction of 1 to 2b, but before phase mixing. Residual 1 is observed in the latter owing to the set SOC. b, Simplified depiction of the captured UO2X2 in the form of 3N and/or 4N. Inset, aqueous UV-Vis spectrum showing the capture of UO2X2 by the 2b/DCE layer (top); the corresponding 31P{1H} NMR spectrum of the DCE layer showing the captured major product (3N/4N) and minor residual 1 (bottom). c, Biphasic release of UO2X2 from the DCE layer to a fresh NaOAc-buffered solution (pH 5.4), following electrochemical oxidation of 3N/4N. Inset, aqueous UV-Vis and organic 31P{1H} NMR spectra of free UO2X2 and 1, respectively—both consistent with the release of captured UO2X2 from the DCE to the aqueous phase. A small amount (~20%) of unknown byproducts (marked by asterisks) is also observed in the 31P{1H} NMR spectrum.Note that exposure to organic solvents would not be acceptable unless the organics were destroyed by subsequent processing. One such available approach to processing would involve subjecting the resultant aqueous solution to supercritical conditions, whereupon the solvent residues would be oxidized to carbon dioxide and the water reduced to hydrogen.
This is a lab scale procedure, and it seems to me that a number of issues need to be addressed before anything like this could be run on an industrial scale. Then again, as stated at the outset, the "breed and burn" concept means that there is really no need to obtain more uranium than has already been mined, at least for several centuries, so there's plenty of time to do that, to make nuclear energy essentially inexhaustible. (At the end of my life, it does seem that ultimately fusion energy may be viable, but current isolated uranium might make the world survivable in the interim.
It's a nice little interesting paper, I think.
Have a nice day tomorrow.
 = new reply since forum marked as read
Highlight:
NoneDon't highlight anything
5 newestHighlight 5 most recent replies
= new reply since forum marked as read
Highlight:
NoneDon't highlight anything
5 newestHighlight 5 most recent replies